When a new disease emerges, scientists around the world take action to figure out what they can do about it, hoping to find new ways to help.
Researchers at the University of Minnesota (UM) have done just that – by examining the structure of the 'spike' protein on the surface of SARS-CoV-2, the team hopes they have contributed to the basis for the development of a new drug.
“In general, by studying which structural features of viral proteins are most important for establishing contact with human cells,” explains biomedical researcher Fang Li, “we can develop drugs that seek them out and block their activity.”
The team used X-ray crystallography to create a 3D model of what the spiky protein looks like and how it binds to human cells.
While this doesn't sound like the pictures of coronavirus you're used to seeing, it's an incredibly useful model for biologists. It allows them to visualize how small mutations in a protein create various folds and ridges that allow the viral particle to attach to receptors in our own cells.
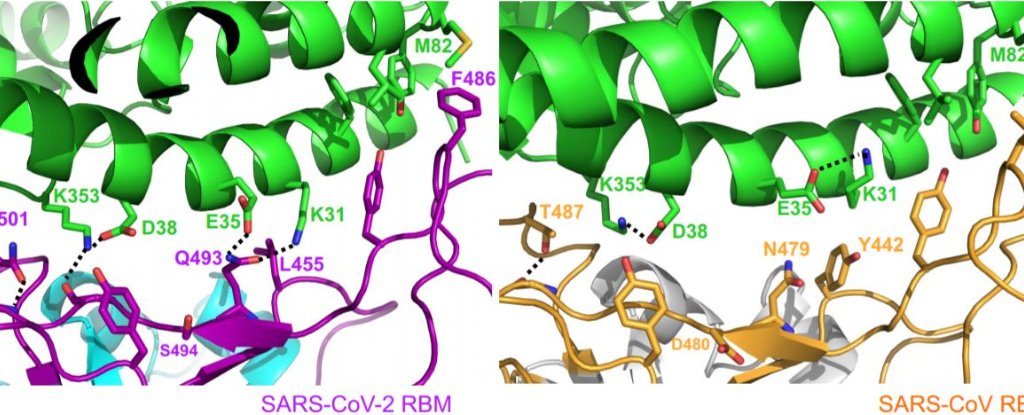
The researchers found that the SARS-CoV-2 coronavirus strain has several mutations that form a particularly compact 'comb' in the spike protein.
This ridge is more compact than that of the SARS virus and may be one of the reasons this new strain is so adept at infecting humans, causing COVID-19.
'The 3D structure shows that compared to the virus that caused the outbreak of SARS in 2002-2003. The new coronavirus has developed new strategies for binding to its human receptor, resulting in tight binding, 'said Li.
“Binding tightly to the human receptor can help the virus infect human cells and spread among humans.”
The team hopes the new simulation will help other researchers develop drugs or vaccines against the virus.
“Our work could help develop monoclonal antibodies that act as a drug to recognize and neutralize the receptor-binding portion of the spike protein,” Lee said.
“Or some of the thorn protein could be the base of the vaccine.”
But we must be careful at this stage. This kind of research is constantly evolving, and although the model is promising, the study used only small fragments of the virus – its binding domain – and therefore, more information remains to be studied.
The study was published in the journal Nature.
Sources: Photo: (Shang et al., Nature, 2020)





